
2020 年 2 月 25 日,北京生命科学研究所/清华大学生物医学交叉研究院黄牛实验室在《Frontiers in Pharmacology》杂志发表题为“Predicting or Pretending: Artificial Intelligence for Protein-Ligand Interactions Lack of Sufficiently Large and Unbiased Datasets”的文章,发现 AI 用于预测蛋白-配体相互作用时,常用的训练集(PDBbind和DUD-E)对AI模型训练存在严重数据偏向(data bias),导致 AI 模型表现虚高,预测缺乏泛化能力和鲁棒性,误导本领域的方法发展和实际应用,并基于此提出了如何客观评估 AI 模型的观点和建议。
近年来,基于神经网络的AI模型多次被宣称在 PDBbind 和/或 DUD-E 蛋白-配体结合数据集上获得了“无与伦比” (state-of-the-art)的表现,预测蛋白-配体结合强弱的能力尤为突出。但是本文作者发现,仅基于配体小分子数据训练AI模型也可以获得同样“无与伦比”“的表现,提示AI模型居然完全不需要学到蛋白-配体相互作用就可以”“预测”蛋白-配体结合能力,这就和药还没到,病就除了差不多。而引起这一悖论的原因是— PDBbind 和 DUD-E 含有数据偏向误导了AI模型。
有一个经典的例子可以说明数据偏向的危害:1957 年,美军支持的一项研究中,研究人员使用神经网络预测树林中是否有坦克,训练集是有坦克或者没坦克的图片,准确度惊人。但是后来发现,有坦克的图片都是在阴天拍摄的,而没坦克的图片都是在晴天拍摄的,这个训练的 AI 模型不是坦克分类器,而是天气分类器!看似复杂的预测坦克存在与否的因果关系学习,被懒惰而又滑头的 AI 用最简单的天气相关性来替代,把人类糊弄其中。偏嗜大数据的AI是动物凶猛的饕餮,并非任人打扮的小菇凉。驯服 AI 首先需要有一个好的训练集,应该在目标属性(如是否有坦克)上与真实分布相符,而在非目标属性(如天气)上无偏向,避免模型学到隐含的数据偏向。

PDBbind 和 DUD-E 并不是为蛋白-配体相互作用预测而专门构建的训练集,它们的主要角色是独立的基准测试集,用于评估模型的预测能力。训练集需要与测试集有区分度,这是基本的常识,也是评估模型可靠性的重要依据。但由于蛋白-配体结合的实验数据匮乏,已报道的AI模型无奈只能在 PDBbind 和 DUD-E 上进行交叉验证(将数据分成 k 分,分别取每一份为测试集,其他 k-1 份为训练集),评估 AI 模型预测蛋白-配体相互作用的能力。这种情况下训练出来的模型可靠性有多大?为了回答这个问题,本文作者针对 PDBbind 和 DUD-E 设计了基线模型(baseline)和交叉验证实验,分析模型会学到哪些隐含的数据偏向。
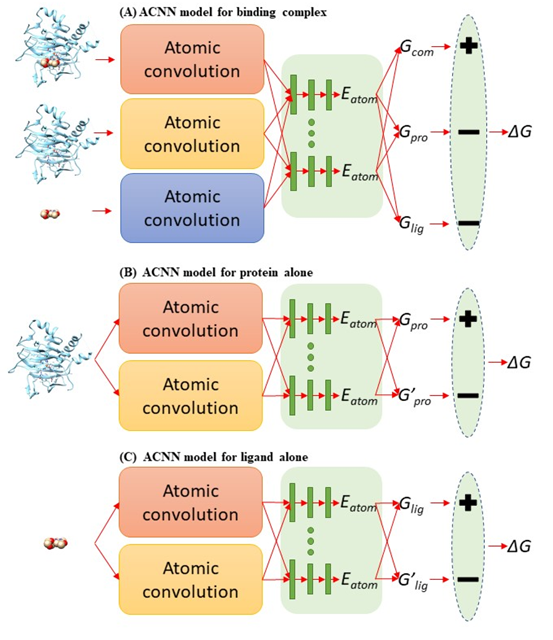
PDBbind 收集了 PDB 蛋白晶体结构数据库中有实验测得结合常数的蛋白-配体复合体,按数据质量(低到高)和数量(大到小)分为 general (11987),refined (3706) 和 core (195) set。本文作者将复合体拆分成蛋白和配体,形成 3 个 PDBbind 数据集: 原版 PDBbind (Binding Complex),只包含配体的 PDBbind (Ligand Alone) 和只包含蛋白的 PDBbind (Protein Alone)。 使用斯坦福大学Pande实验室发展的原子卷积神经网络(ACNN)模型,在 refined set 或 general set上训练蛋白-配体相互作用力预测模型,训练集中都去除了 core set, 然后预测 core set中复合物的蛋白-配体相互作用强弱,ACNN 的表现见下图。可以看到,仅使用蛋白或者仅使用配体结构作为输入来计算蛋白-配体相互作用强弱,就能获得与复合体作为输入相近甚至更好的表现,可以说是 PDBbind v2015 core set 上的“state-of-the-art”,在 PDBbind v2018上表现也类似。该结果揭示—AI模型居然无需学到蛋白-配体相互作用模式就可以计算蛋白-配体相互作用?这一违反常理的现象只能反推得到一个合理的解释 — 采用 PDBbind 数据训练AI模型有严重偏向性,简而言之,对饕餮 AI 而言,PDBbind 数据集吃不饱(数据量不够大),还严重偏食(多样性不够高)。
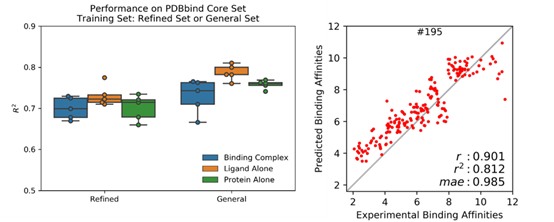
通过进一步限制训练集和测试集间的配体小分子骨架(scaffold)或者蛋白序列的相似度, ACNN 模型的表现有明显下降,说明模型可能通过简单的配体相似度或者蛋白相似度”预测”蛋白-配体相互作用,即类似的配体有类似的结合活性,类似的蛋白也有类似的结合活性,而无需学到复杂的蛋白-配体结合模式。两点之间,AI 永远会走最短的直线。这体现了神经网络强大的拟合能力,而且善于发现相关关系。但是,这样的模型只在与训练集非常近似的场景中才能准确预测,难以泛化,需要海量且多样的数据才能构建一个鲁棒的 AI 模型。在蛋白-配体复合物晶体结构和活性测定数据匮乏且昂贵的领域,这个问题显得更难以克服。因此,当这些模型面对与训练集天差地别的真实世界复杂的药物发现和优化的情景时,纸上谈兵感呼之欲出。
既然实验来源的数据不够,那是不是可以用计算机生成的数据来训练模型?DUD/DUD-E 作为常用的分子对接基准测试集,不仅包含实验测定的 22886 个活性小分子(active),还包括 141 万类药小分子作为阴性对照(decoy)。Decoy 需要和active有相似的理化性质(分子量,净电荷等),但不同的拓扑结构(以分子指纹表征)减少假阴性。它克服了之前基准测试集的缺点,即分子对接软件仅通过简单的理化性质就能分辨active和decoy,在基准测试集中获得高分。由此可见,传统的分子对接同样掉进过陷阱(仅依赖简单的理化性质打分排名),但通过设置合理的阴性对照爬了出来,有了可靠的测试集用于客观评估方法的好坏。但AI的难题在于我们知道它的陷阱是什么吗?数据集的相似性?数据集的大小?为了回答这个问题,在数据量更大的DUD-E数据集训练AI模型,分别以 6 种理化性质(PROP)和分子指纹(FP)作为输入特征,训练随机森林(RF)对 active 和 decoy 进行分类。
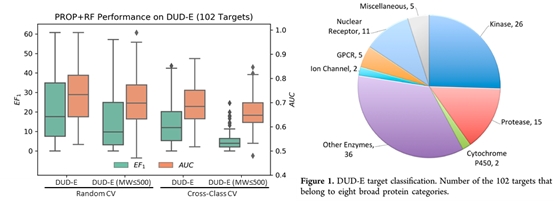
当使用 6 种理化性质为输入特征,如果随机分为 3 组做交叉验证(CV),随机森林在DUD-E 102个靶点的平均 AUC 为0.73,平均前 1% 活性小分子富集系数(EF1)为 22.2,非常接近 DUD-E 文章中分子对接软件的表现 (AUC: 0.76, EF1: 19.8)。在去除分子量大于 500 的小分子(已报道有偏向),并且按蛋白类型(class)进行分组交叉验证,AUC 降为 0.66,EF1 降为 5.14,说明在 DUD-E 上训练的模型有可能学到理化性质上的偏向。包括: 1) active中含有分子量大于 500 的小分子,而 decoy 由于限定为类药小分子,分子量都小于500; 2) 同类靶点的 active 的理化性质相似,模型可以仅通过理化性质区分 active 和 decoy。
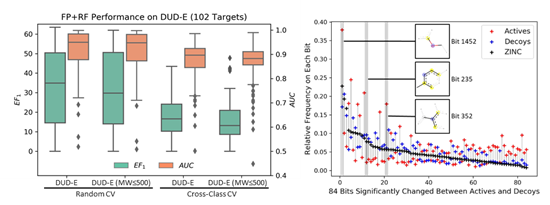
当使用分子指纹为输入特征,即使在难以学到理化性质偏向的情况下,随机森林模型也能很好地区分 active 和 decoy。将分子指纹中高频且在 active 和 decoy 中差异出现的84个特征,以在 ZINC 数据库中的出现频率排序 (见上图),可以发现DUD-E 在拓扑结构(分子指纹)上存在偏向主要有两个原因:1) DUD-E是从ZINC中挑选与active拓扑结构不相似的小分子作为decoy,所以active和decoy和预期一样有明显差异;2) Decoy和ZINC 的分布更接近,说明 active 和 ZINC 的拓扑结构分布本就不同。DUD-E 在理化特性和拓扑结构上都有偏向,只要模型可以显式或者隐式地学到这些特征,即使基于对接的复合体训练模型,也难以避免被偏差误导。
综上,作者认为现阶段缺乏充足且无偏的数据用于训练基于蛋白-配体复合物结构的 AI 药物发现和设计模型。由于AI模型强大的总结相关性的能力,为了合理评估AI模型对蛋白-配体结合强弱的预测能力,促进该领域地健康发展,提出以下建议:
- PDBbind 仍将是目前为止最合适的实验数据集。但在使用 PDBbind 训练模型时,应该设置 protein alone 和 ligand alone 模型作为基线对照,以恰当评估模型提升的原因。
- 应该系统地控制训练集和测试集间的蛋白相似度和配体相似度,以恰当评估模型的泛化能力。
- DUD-E 数据集应该作为独立的基准测试集,而不是训练集。