

在图像识别领域,ImageNet数据集的重要性不言而喻。在生物计算领域,2024年黄牛实验室发布的BindingNet v1数据集独树一帜,采用模板匹配技术构建了高质量的蛋白-配体复合物三维结构模型,有效减轻了AI模型在蛋白-小分子亲和力预测中因包埋溶剂可及表面积引发的偏见等问题,为蛋白-配体相互作用研究领域中AI模型的评估和优化奠定了坚实基础,此阶段我们称之为“From Big Data to Good Data”。
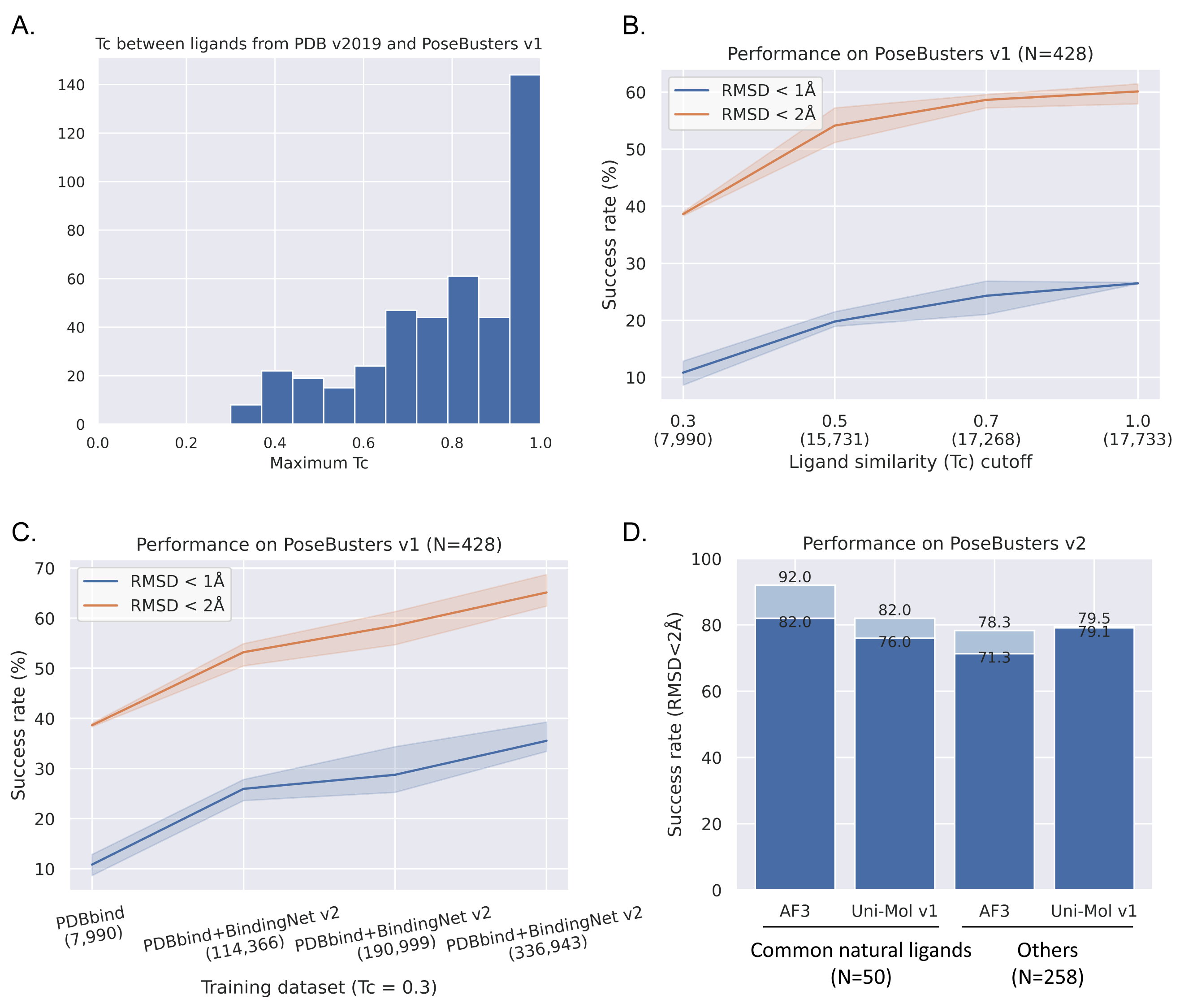
然而,AI对数据的需求永无止境,接下来我们进入 “Make Good Data Greater”的阶段。2025年1月8日,北京生命科学研究所/清华大学生物医学交叉研究院黄牛实验室于《npj Drug Discovery》杂志在线发表题为“Augmented BindingNet dataset for enhanced ligand binding pose predictions using deep learning”的研究论文。在BindingNet v1数据集的模板匹配技术基础上,创新性地提出了基于片段化结合形状与静电匹配的多层次模板匹配流程,成功构建出规模远超BindingNet v1近十倍规模的蛋白-配体相互作用数据集— BindingNet v2。此数据集覆盖1794个蛋白靶点,包含689,796个蛋白-小分子配体复合体结构模型及其相应的实验活性数据,极大地提升了AI模型在蛋白-配体复合体预测任务上的泛化能力。尤其在仅使用Tc < 0.3的数据(不包含同测试集中结构类似的分子)训练时,Uni-Mol模型在PoseBusters测试集上的泛化能力从38.55%大幅跃升至74.07%。
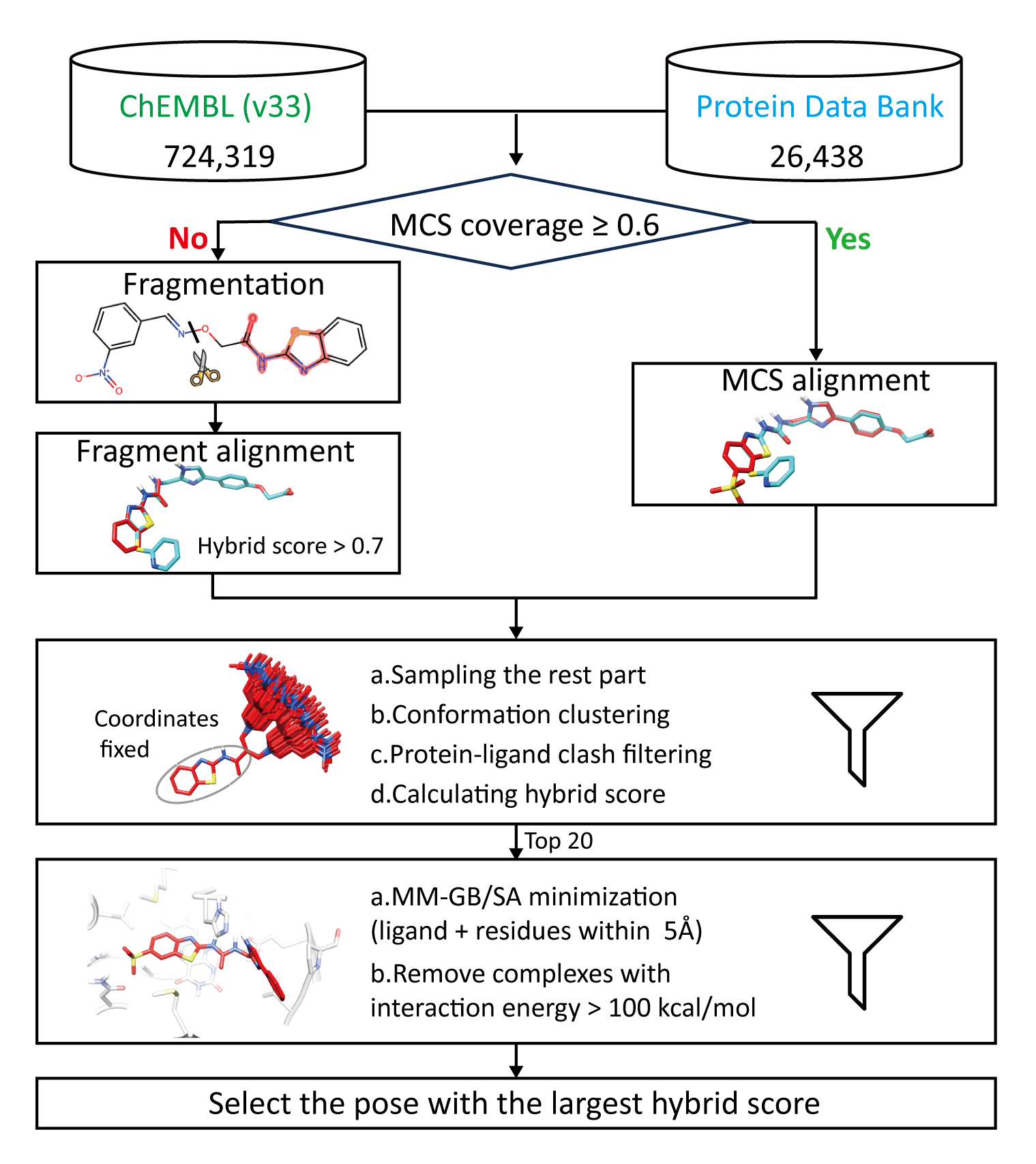
多层次的模板匹配流程
研究团队从 PDB 数据库中筛选出 26,438 套高质量的蛋白-小分子复合物结构作为模板,并从 ChEMBL 数据库中挑选出 724,319 对实验验证的蛋白-小分子数据对。随后,通过以下五个步骤构建多层次模板匹配流程(图1):
- 计算候选分子与模板分子之间的最大公共子结构占有率。
- 关键子结构叠合:
- 若候选分子的最大公共子结构占有率超过 0.6,则直接将候选分子与模板分子进行叠合;
- 若未达到占有率要求,则对候选分子进行片段化处理,并借助SHAFTS 工具探寻片段与模板分子间三维形状及药效团叠合程度(即 hybrid score)最高的构象。
- 构象采样:使用ETKDG对叠合后的剩余部分结构进行采样、聚类、过滤操作,计算hybrid score,以确保构象的合理性。
- 能量最小化:挑选hybrid score排名前20个的复合物结构进行MM/GB-SA优化。
- 打分:选取 hybrid score 最高的复合体作为最终构象。
BindingNet v2数据集与网站介绍
借助多层次的模板匹配流程,研究团队成功构建了689,796个蛋白-小分子复合物结构,并为每个复合物附上对应的实验活性数据。同时,依据 hybrid score 对数据集进行质量分级,其中高质量(hybrid score ≥ 1.2)、中质量(1.0 ≤ hybrid score < 1.2)、低质量(hybrid score < 1.0)的数据分别占33.63%、23.91%、42.45%。为便于科研人员检索和分析,团队搭建了专属网站,用户可通过该网站查看构建的复合物三维结构,开展蛋白-小分子相互作用分析。
BindingNet v2对深度学习模型泛化能力的显著提升
尽管已有多个深度学习模型在 PoseBusters 测试集上展现较高的成功率(如 Uni-Mol v1:62.4%、AlphaFold3 with pocket specified:90.0%),但本研究团队发现,这些模型所采用的训练集与测试集之间小分子结构存在很高的相似性。例如,测试集中70.09%的样本能在PDB(v2019)中找到相似性大于0.7的训练样本,超过90%的测试集样本都能从PDB(v2019)中找到相似性大于0.3的训练样本。然而,在实际应用中,基于靶标结构的虚拟筛选最有意义的应用场景是发现全新骨架的小分子,且通常以 Tc < 0.3 作为最严格的筛选标准。因此,研究团队仅使用Tc < 0.3的训练样本严格评估Uni-Mol的泛化能力。结果显示,仅用 PDBbind(Tc < 0.3) 数据训练时,Uni-Mol 模型在 PoseBusters 测试集上的成功率仅为 38.55%;随着BindingNet v2 中Tc < 0.3 的数据逐步加入训练集,模型泛化能力显著提升至 64.25%;结合MM/GB-SA优化和重打分后,成功率进一步提升至 74.07%,并通过了所有物理化学合理性检查(图2)。值得关注的是,仅用 Tc < 0.3 数据训练的模型在类药子集上的成功率已超越用PDB(v2019)训练的AlphaFold3模型,充分验证了BindingNet v2 数据集的重要价值。然而,BindingNet v2中的蛋白和小分子结构多样性仍然受限于PDB数据库,未来,黄牛实验室将通过整合多层次数据、深度学习预测复合体结构、结合物理方法优化结构、以及引入半自动化质量标注流程等多方面的迭代优化,为蛋白-配体相互作用研究领域提供更全面且高质量的数据支持,进而提升人工智能方法在柔性对接、复合体构象动态过程预测以及蛋白-小分结合能预测等方面的表现。
黄牛实验室的博士研究生朱慧为本文第一作者,黄牛博士为通讯作者。其他作者包括黄牛实验室的李雪莲博士和陈保全。ByteDance AML – AI for Science Team为本研究提供了重要的算力支持。该项研究获得北京市科委和清华大学资助,在北京生命科学研究所完成。此外,特别感谢李伟博士对本文相关内容撰写方面给予的重要帮助。