
Xiaobo Wan,†,‡§ Wei Zhang,‡§ Li Li,‡ Yuting Xie,‡ Wei Li†,‡ and Niu Huang*,‡,†
† Graduate School of Peking Union Medical College and Chinese Academy of Medical Sciences,Beijing, China
‡ National Institute of Biological Sciences, Beijing, No. 7 Science Park Road, Zhongguancun Life Science Park, Changping District, Beijing 102206, China
§These authors equally contributed to this work.
* To whom correspondence should be addressed. Phone: 86-10-80720645. Fax: 86-10-80720813.
E-mail: huangniu@nibs.ac.cn
ABSTRACT: The rational design of selective kinase inhibitors remains a great challenge. Here
we describe a physics-based approach to computationally modeling the kinase inhibitor
selectivity profile. We retrospectively assessed this protocol by computing the binding profiles of
17 well-known kinase inhibitors against 143 kinases. Next, we predicted the binding profile of
the chemotherapy drug mitoxantrone, and chose the predicted top five kinase targets for in vitro
kinase assays. Remarkably, mitoxantrone was shown to possess low nanomolar inhibitory
activity against PIM1 kinase and to inhibit the PIM1-mediated phosphorylation in cancer cells.
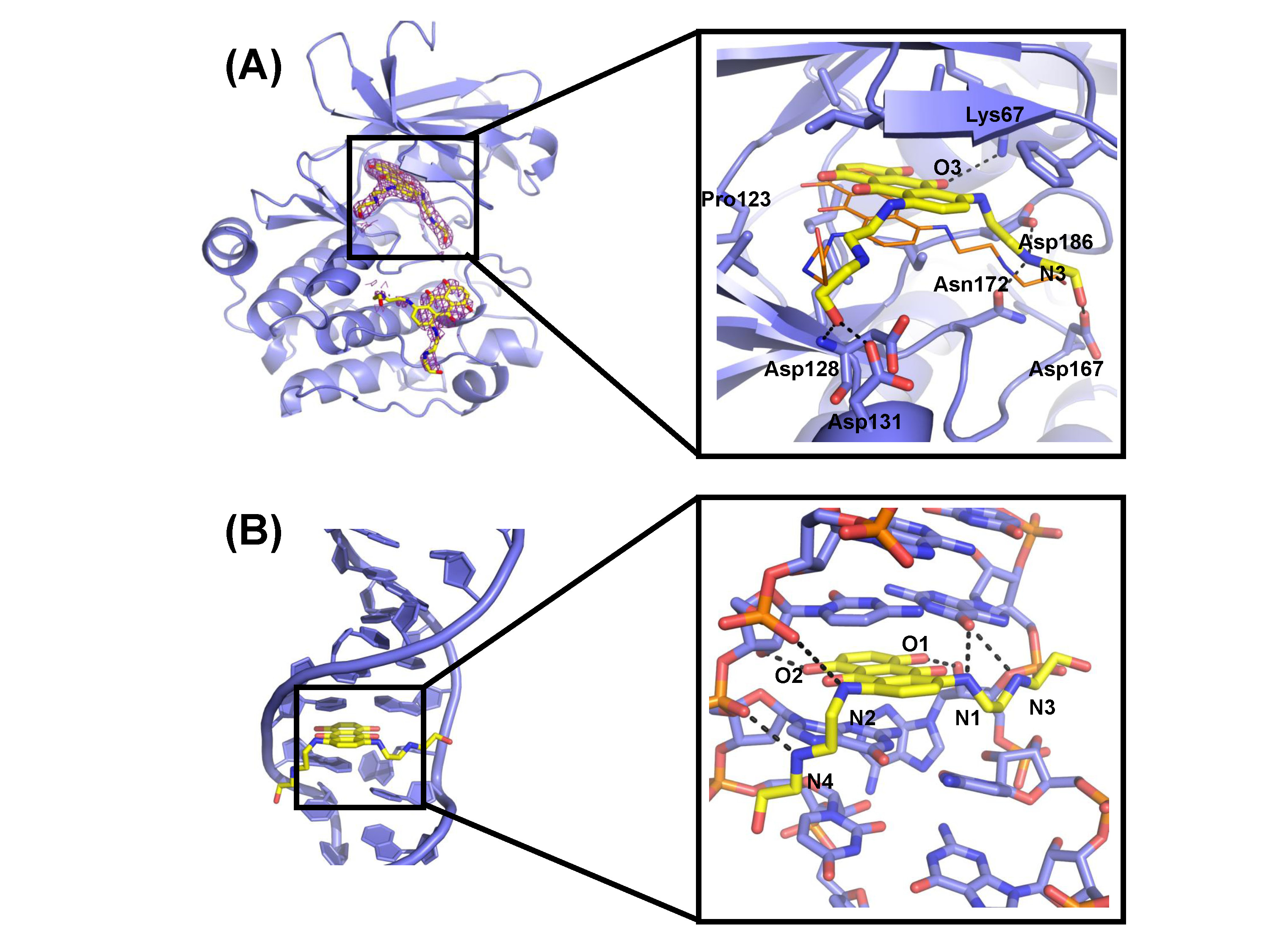
We further determined the crystal complex structure of PIM1 bound with mitoxantrone, which
reveals the structural and mechanistic basis for a novel mode of PIM1 inhibition. Although
mitoxantrone’s mechanism of action had been originally thought to act through DNA
intercalation and type II topoisomerase inhibition, we hypothesize that PIM1 kinase inhibition
might also contribute to mitoxantrone’s therapeutic efficacy and specificity.