

From Big Data to Good Data: 利用模板匹配方法构建高质量的蛋白-配体复合物结构模型数据集BindingNet
“Content without method leads to fantasy; method without content to empty sophistry.”— Goethe
2024年热闹非凡的JPM大会已经结束,前几年锣鼓喧天的AI制药公司显得冷清了许多。AI加速新药发现提高新药研发成功率,是啤酒花还是泡沫,正在经历Hype Cycle低 谷期的行业中人,可能对这个问题会有更深刻的认识。是AI本身不行吗?不是的,众所周知,AI在图像识别和生成领域非常成功。回顾其发展历程,会发现2009年是一个特别的年份,这一年ImageNet横空出世,”ImageNet改变了AI领域人们对数据集的认识,人们真正开始意识到它在研究中的地位,就像算法一样重要”,李飞飞教授说。如今大家意识到,在AI落地具体场景时,数据可能比算法更加重要。
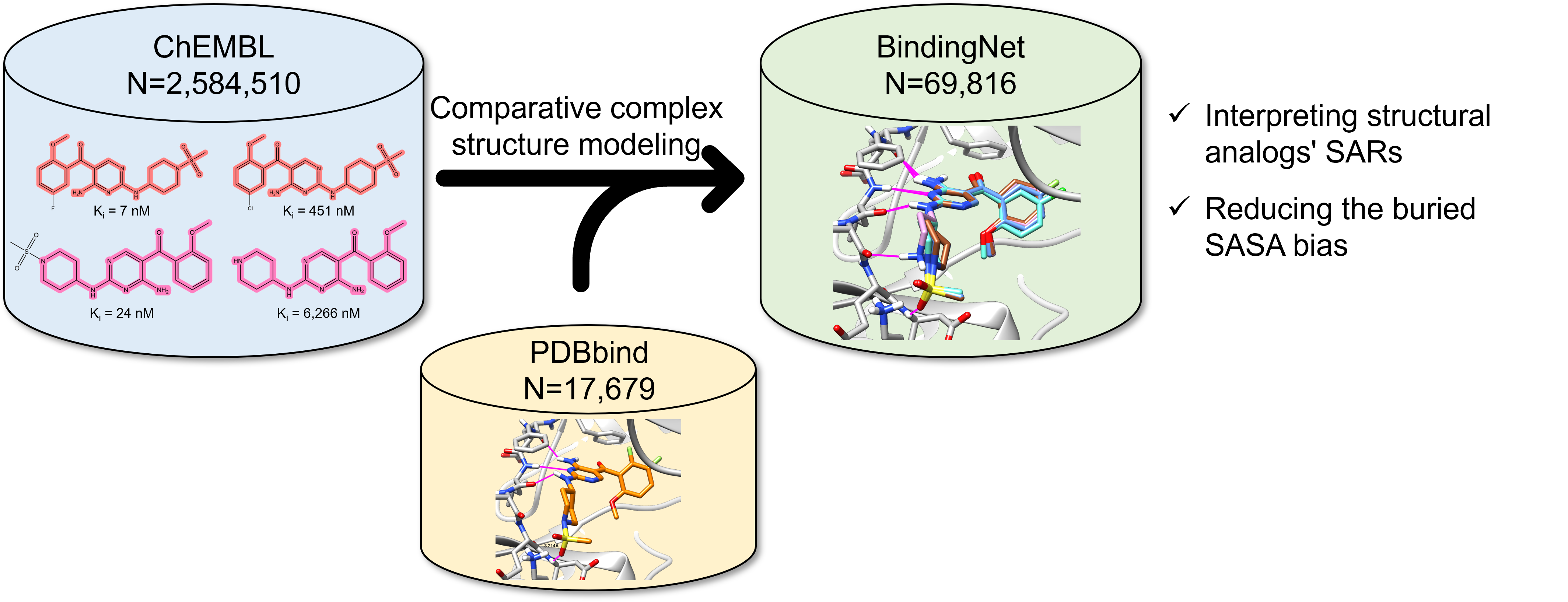
AI加速新药发现的一个基石问题是,AI能否准确预测蛋白-配体结合亲和力?然而,现有的蛋白-配体复合物实验结构数量和类型非常有限,不足以用于训练有泛化能力的AI模型。而unfair datasets又让我们高估AI模型的预测能力,以至于在前瞻性的新药开发实践中失去作用。工欲善其事,必先利其器。Pat Walters甚至直接呼吁“As a field, we must reach a consensus on appropriate datasets and statistical tests for method comparisons”。类比ImageNet,AI制药领域需要构建数量足够、数据清晰、类型多样化的数据集,对于正确评估和优化提高AI模型具有重要意义。为此,北京生命科学研究所/清华大学生物医学交叉研究院的黄牛实验室利用模板匹配方法构建了一个包含69,816个高质量蛋白-配体复合物模型和相应的实验结合活性数据的BindingNet数据集,作为领域内最常用数据集PDBbind的补充。作者探索了利用BindingNet数据集进行结构活性关系(SAR)分析的潜在应用,研究了基于BindingNet训练的深度学习模型在预测蛋白-配体结合亲和力方面的性能。发现基于BindingNet训练的深度学习模型可以减轻由包埋的溶剂可及表面积(buried SASA)引起的偏见。 “不积跬步,无以至千里”,如何进一步完善BindingNet数据集,扩大其覆盖的化学空间和蛋白-配体对的种类将是下一步工作的重点。近日,该项研究工作发表在J. Chem. Inf. Model 的Machine Learning in Bio-cheminformatics专刊中【1】。
BindingNet的构建方式:
- 以PDBbind v2019数据集中的小分子配体为模板,从ChEMBL数据库中搜寻与其同一靶标的结构类似的系列活性分子(相似度大于70%),共找到5907个PDBbind模板结构作为候选的匹配对象。
- 利用最大公共子结构(MCS)模板匹配的方式构建初始的复合物结构,并通过对非公共部分的构象搜索和配体分子结合构象的MM/GB-SA优化和打分,来保证复合物结构模型的可靠性,共生成合格的高质量蛋白-配体复合物结构模型69,816套。
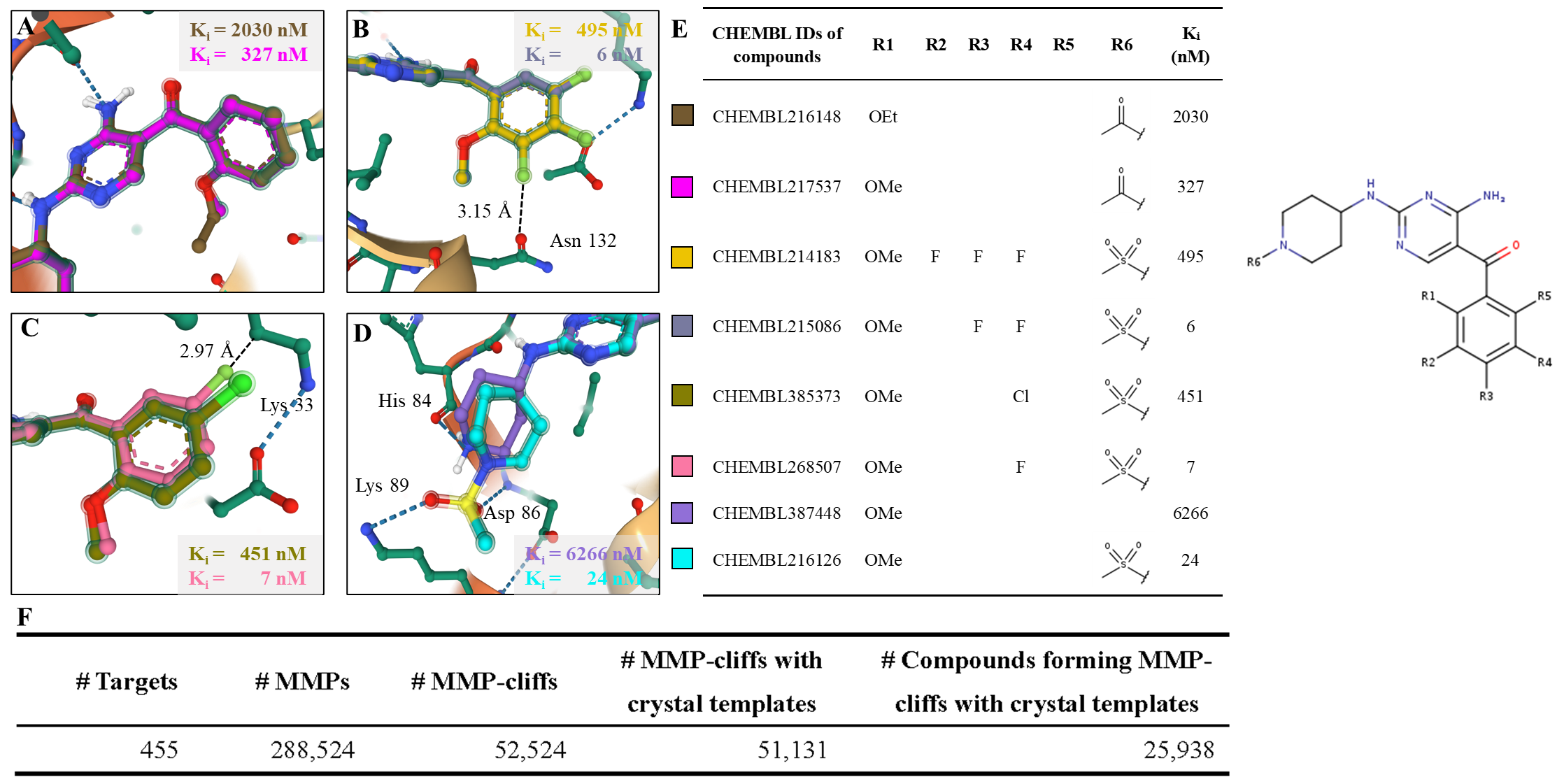
SAR分析和活性悬崖分子对数据:
利用这种方式构建的BindingNet包含了丰富的结构活性关系(SAR)信息,即同一蛋白靶标与不同小分子的结合构象和活性的变化。其中还包含了许多活性悬崖分子对(MMP-cliffs),即结构变化微小但活性差异显著的分子对,这些分子对有助于理解蛋白-配体相互作用的关键因素。例如,通过分析BindingNet提供的CDK2-抑制剂复合物的模型,可以合理地解释活性变化的具体原因,如在R2位点引入F原子会增加其与Asn132羰基氧之间的静电排斥,导致活性下降近100倍。为了方便用户查询、分析和下载BindingNet数据集,作者提供了免费公开的网站http://bindingnet.huanglab.org.cn。
机器学习模型开发和评估:
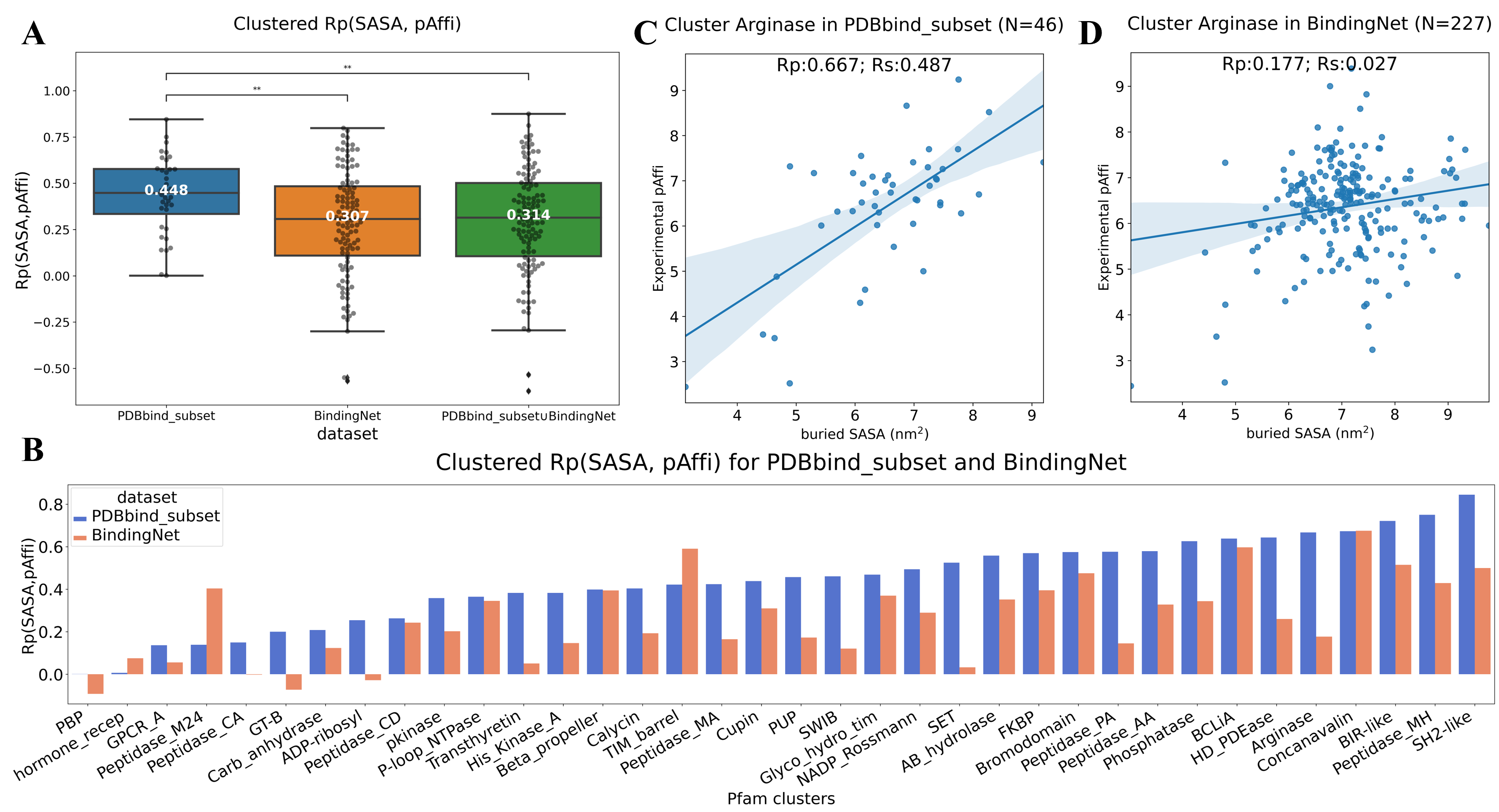
BindingNet数据集可用于开发和评估机器学习模型,预测蛋白质-配体复合物结合活性、结合位置以及分子生成等任务。在蛋白-配体相互作用预测方面, PDBbind是最常用的训练集。但有研究指出PDBbind作为训练集存在的一些问题,如数据量不足、过于稀疏等【2】。此外,该研究小组还曾报道,由PDBbind训练的机器学习模型对包埋溶剂可及表面积(buried SASA)存在一定的偏见,即buried SASA 越大,模型预测的蛋白-小分子结合活性越好【3】。由于BindingNet包含大量结构相似,但结合活性差异显著的小分子,作者推测基于BindingNet训练的机器学习模型能减轻先前在PDBbind训练的模型中发现的buried SASA偏见。因此在PDBbind_hold_out_2019 set上进行测试,发现基于BindingNet_v18训练的模型确实比基于PDBbind_v18_subset训练的模型表现差,且PDBbind_v18_subset的复合物模型预测的结合活性与buried SASA相关性更好,Rp达到了0.623。进一步的分析发现训练集PDBbind_v18_subset和测试集PDBbind_hold_out_2019数据集中的复合物结合活性与buried SASA本身就具有一定的相关性,而 BindingNet_v18数据集中该相关性更弱。作者统计了PDBbind_subset和BindingNet中每个蛋白家族内的Rp(SASA,pAffi),证实了BindingNet数据集中的Rp(SASA,pAffi)明显低于PDBbind_subset。
总结与展望:
综上所述,利用配体模板匹配的方法扩充PDBbind数据集,是个在实验结构数据稀疏的情况下的可行方案。BindingNet可以为药物化学家在系列类似物的晶体结构尚未确定的情况下,在原子水平上研究蛋白-配体相互作用提供有益的见解。作者发现基于BindingNet训练的机器学习模型可以减轻先前在PDBbind上发现的buried SASA偏见,这得益于BindingNet规模更大且包含了大量的buried SASA相似但结合活性不同的复合物结构数据。BindingNet数据集也可用于开发和评估结合位置预测、配体结合自由能计算和活性悬崖预测等方法。作者提出还需要进一步努力来完善BindingNet数据集的构建,以扩大其覆盖的化学空间和蛋白-配体对的种类。可以考虑整合多种方法,如分子对接和基于机器学习的方法,来预测不同靶标的蛋白-配体复合物的结合位置和增加小分子多样性。为了扩大覆盖范围,还可以考虑包括不同的生物活性数据库,如BindingDB和Binding MOAD,以及PDBbind未收录的复合物实验结构。在AI制药领域构建出行业认可和适用的“ImageNet”,BindingNet只是一个起点,但希望也能成为AI制药进入Hype Cycle另一个阶段的起点。
【1】Li, X.; Shen, C.; Zhu, H.; Yang, Y.; Wang, Q.; Yang, J.*; Huang, N*. A High-Quality Data Set of Protein–Ligand Binding Interactions Via Comparative Complex Structure Modeling. J. Chem. Inf. Model. 2024.
【2】Yang, J.; Shen, C.; Huang, N*. Predicting or Pretending: Artificial Intelligence for Protein-Ligand Interactions Lack of Sufficiently Large and Unbiased Datasets. Front. Pharmacol. 2020, 11, 69.
【3】Zhu, H.; Yang, J.*; Huang, N*. Assessment of the Generalization Abilities of Machine-Learning Scoring Functions for Structure-Based Virtual Screening. J. Chem. Inf. Model. 2022, 62 (22), 5485–5502.